
Drug Repurposing
The development of herbal formulation in the current scenario for the prevention and treatment of COVID-19 has been possible because of the uses of miraculous molecular docking studies of the phytoconstituents…
Dr Kali Charan Sharma and Dr Mahaveer Dhobi


A countless use of drug repurposing is to identify drugs that were developed for treating other diseases to treat a new disease. Drug repurposing can be accomplished by conducting systematic drug target interaction (DTI) analyses. It is important to identify potential DTIs for both approved drugs, natural compounds and drug candidates, which serves as the basis of repurposing drugs and selection of drug targets without DTIs that may cause side effects.
 The computational approaches such as computer-aided virtual screening and molecular modelling provide a significant breakthrough in understanding the structural aspects of various molecular targets of coronavirus and identification novel lead molecules. Natural molecules are one of the probable alternatives as many of these compounds possess ideal drug likeliness and pharmacokinetic features and might probably be used as lead molecules against various targets of SARS-CoV-2. Our study provides acomputational strategy to combat SARS-CoV-2 and its genetic and structural information, conventional drugs currently used and their shortcomings, various anti-viral compounds present in nature, computational approaches for molecular modelling of the target proteins, major drug targets that are identified, and virtual screening of herbal-based molecules using molecular docking and molecular dynamic simulation studies. Our study focused on portraying the relevance of utilizing natural lead molecules by virtual screening and ADMET prediction for the development of effective lead molecules against SARS-CoV-2.
The computational approaches such as computer-aided virtual screening and molecular modelling provide a significant breakthrough in understanding the structural aspects of various molecular targets of coronavirus and identification novel lead molecules. Natural molecules are one of the probable alternatives as many of these compounds possess ideal drug likeliness and pharmacokinetic features and might probably be used as lead molecules against various targets of SARS-CoV-2. Our study provides acomputational strategy to combat SARS-CoV-2 and its genetic and structural information, conventional drugs currently used and their shortcomings, various anti-viral compounds present in nature, computational approaches for molecular modelling of the target proteins, major drug targets that are identified, and virtual screening of herbal-based molecules using molecular docking and molecular dynamic simulation studies. Our study focused on portraying the relevance of utilizing natural lead molecules by virtual screening and ADMET prediction for the development of effective lead molecules against SARS-CoV-2.
Jenwitheesuk and Samudrala completed a study that used a protein-inhibitor docking approach against HIV Protease1 to find out saquinavir and nelfinavir. HIV-1 protease has special flaps that are in motion upon binding. Since the structure of target protein is rigid, the opening and closing of the flaps is not performed. This protocol was used to simulate the flexible nature between the ligand and the enzyme.
Target Identification and Molecular Modelling Methodology:
In this work, we applied multiscale modelling techniques to identify drugs that may be repurposed to target SARS-CoV-2 ACE2,ORF8 and RdRp target receptor. Three proteins ACE2 (PDB ID: 2AJF), ORF8 (PDBID: 6BB5)and RdRp (PDBID: 7BV2) were selected for docking protocols for identifying SARS-COV-2 ligands.
Phytoconstituents molecules were docked using the GLIDE module of Maestro 9.6 as per the Glide protocol given in Schrödinger. All the default parameters were used. For ligand preparation, the pH was 7.0 ± 2.0, the force field was OPLS3, and ionization was done using Epik. For protein preparation, the pH was 7.0 ± 2.0, the force field was OPLS3, ionization had done using Epikand.
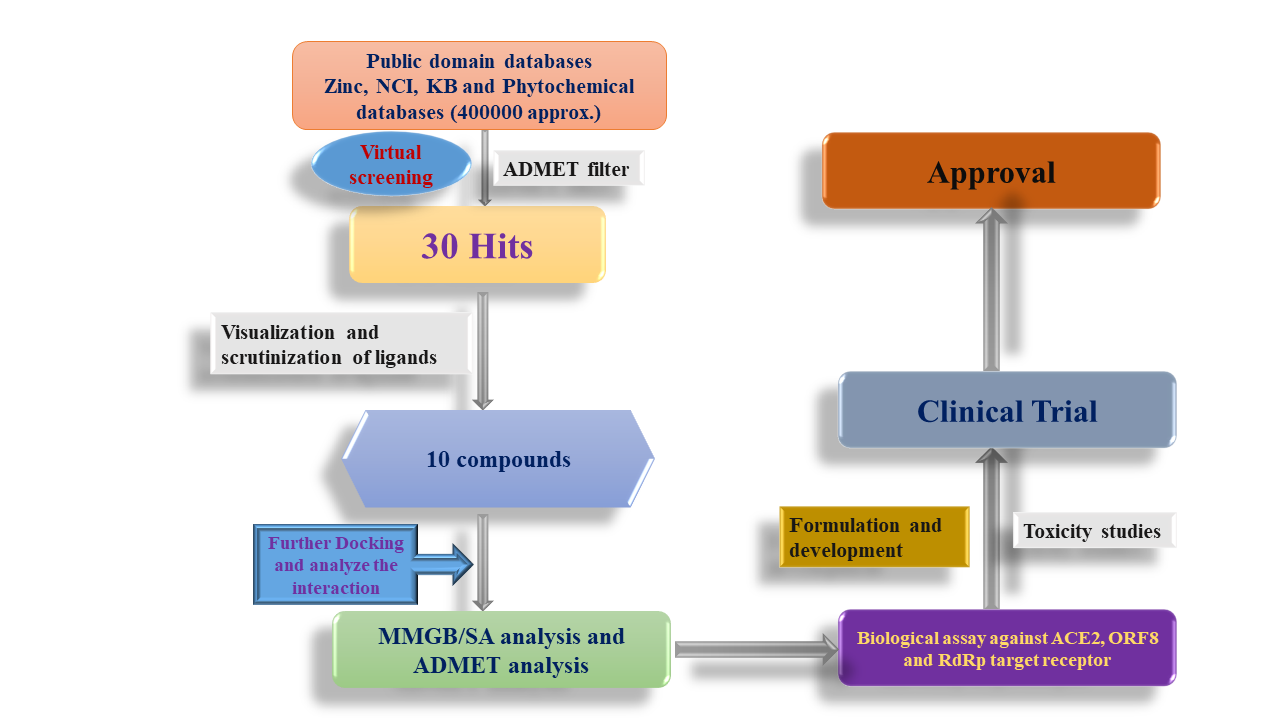
 A total of 400000 compounds synthetic or natural origin were docked to the binding site of five proteins ACE2 (PDB ID: 2AJF), ORF8 (PDB ID: 6BB5)and RdRp (PD BID: 7BV2) with Glide docking module of Schrödinger. During Virtual screening, we put the ADMET filter to remove undesired compounds. The docked poses were then analyzed using Schrödinger software’s imaging tools. After Virtual screening we have got 10 hits which were again go for MMGB/SA analysis and ADMET for selected compounds.
A total of 400000 compounds synthetic or natural origin were docked to the binding site of five proteins ACE2 (PDB ID: 2AJF), ORF8 (PDB ID: 6BB5)and RdRp (PD BID: 7BV2) with Glide docking module of Schrödinger. During Virtual screening, we put the ADMET filter to remove undesired compounds. The docked poses were then analyzed using Schrödinger software’s imaging tools. After Virtual screening we have got 10 hits which were again go for MMGB/SA analysis and ADMET for selected compounds.
Schematic Representation of Molecular Modelling Strategy
Molecular Docking Studies of Various Phytoconstituents with ACE2, RdRp And ORF8
Since the outbreak of COVID-19 pandemic, continuous attempts are being made worldwide to develop a vaccine and utilize repurposing approaches to find out an effective cure and preventive measure. Drug development using herbal drugs which are in use for the treatment of respiratory viral infections and act as immunomodulators is no exception to this. In these directions, two types of targets are mainly exploited; first is SARS-CoV-2 proteins to prevent the entry of virus in host cells to reduce viral load, and second is to modulate human proteins to control the replications of virus and/or to counter the immune-pathogenicity. With continuous research, a lot has been known about the COVID-19 right from SARS-CoV-2 entry in human beings till the severity of pathology it imposes, and mechanism involved therein.
SARS-CoV-2 binds to ACE2 on the host cell membrane and, by endocytosis, enters into the cell. Small molecules targeting the viral binding to the host cells, namely ACE2 in SARS-CoV-2, and nucleoside analogues which interfere with viral replication, are potential therapeutics for treating viral diseases. There are no known viral entry blockers that target host ACE2. Blocking viral entry into host cells may be the most effective strategy for prophylactic and therapeutic treatment of COVID-19.

The natural compounds, especially from plant resources, remained the choice in the lead identification programs due to their abundance, safety and broad spectrum of ensuing activities. The natural products (esp. flavonoids, bioflavonoids or polyphenolic compounds) are a group of secondary metabolites present in fruits and vegetables and are well known for their health benefits. Some of the active phytoconstituents in medicinal plants have inhibitory action on coronaviruses, however determination of functional actions on ACE2 is critical to elucidate agonist/activation or inhibitor/antagonist.
We have attempted to identify naturally occurring phytoconstituents that may target ACE2 and block viral entry using molecular docking studies. We also evaluated the molecular interactions of a selected number of naturally occurring phytoconstituents at host AT1 and AT2 receptors, and SARS-CoV-2 RdRp and ORF8 proteins using molecular docking.
Identification of Medicinal Plants for the Selection of Potential Phytoconstituents through Molecular Docking Studies
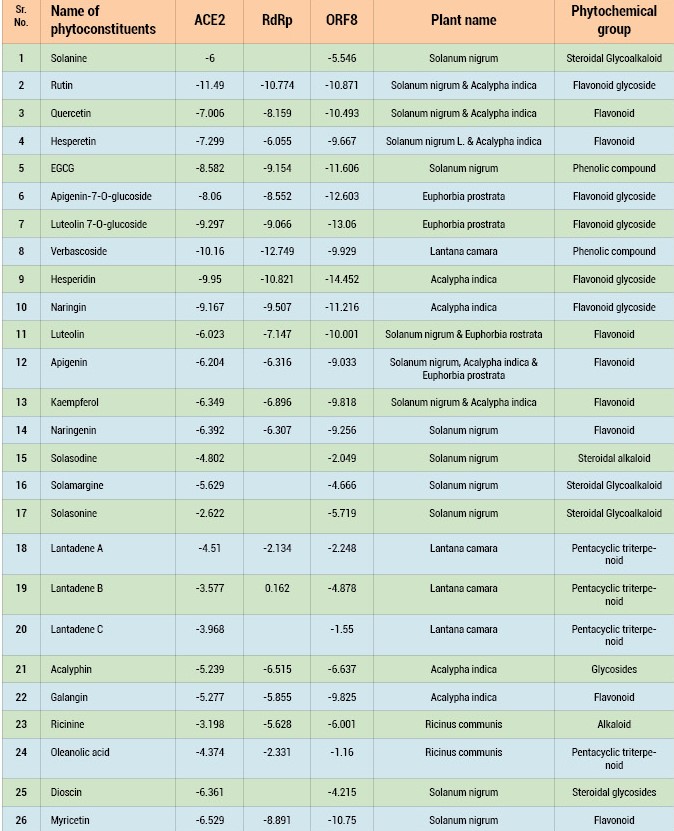
We identified some Indian medicinal plants with potent anti-inflammatory, immunomodulatory, and antiviral activities for developing potential treatment of COVID-19. These plants identified were Solanum nigrum, Euphorbia prostrata, Acalypha indica, Lantana camara and Ricinus communis. A literature search to identify the phytoconstituents of these plants revealed that they contain flavonoids, flavonoid glycosides, triterpenoids, steroidal glycoalkaloids etc. Flavonoids, in particularly, luteolin, quercetin, apigenin, amentoflavone, epigallocatechin (EGC), epigallocatechin gallate (EGCG), gallocatechin gallate (GCG) and kaempferol were reported to inhibit the proteolytic activity of SARS-CoV 3CLpro and 3a channel protein of coronavirus. Certain flavonoids viz., luteolin, quercetin, rutin, kaempferol, and apigenin showed ACE inhibitory activity. In addition to flavonoids, we have also docked certain other compounds of phytochemical groups like triterpenoids, steroidal glycoalkaloids, phenolic compounds since these were major chemical constituents of the antiviral plants mentioned above. Molecular docking study was also carried out for selected steroidal glycoalkaloids: solamargine, solasonine, solasodine, solanine; flavonoids: rutin, naringenin, luteolin, myricetin, quercetin, apigenin, kaempferol and hesperetin, hesperidin, apigenin-7-O-glucoside, luteolin-7-O-glucoside; triterpenoids: Lantadene A, B, C, oleanolic acid, acalyphin, ricinine, dioscin, and epigallocatechin gallate, verbascoside.
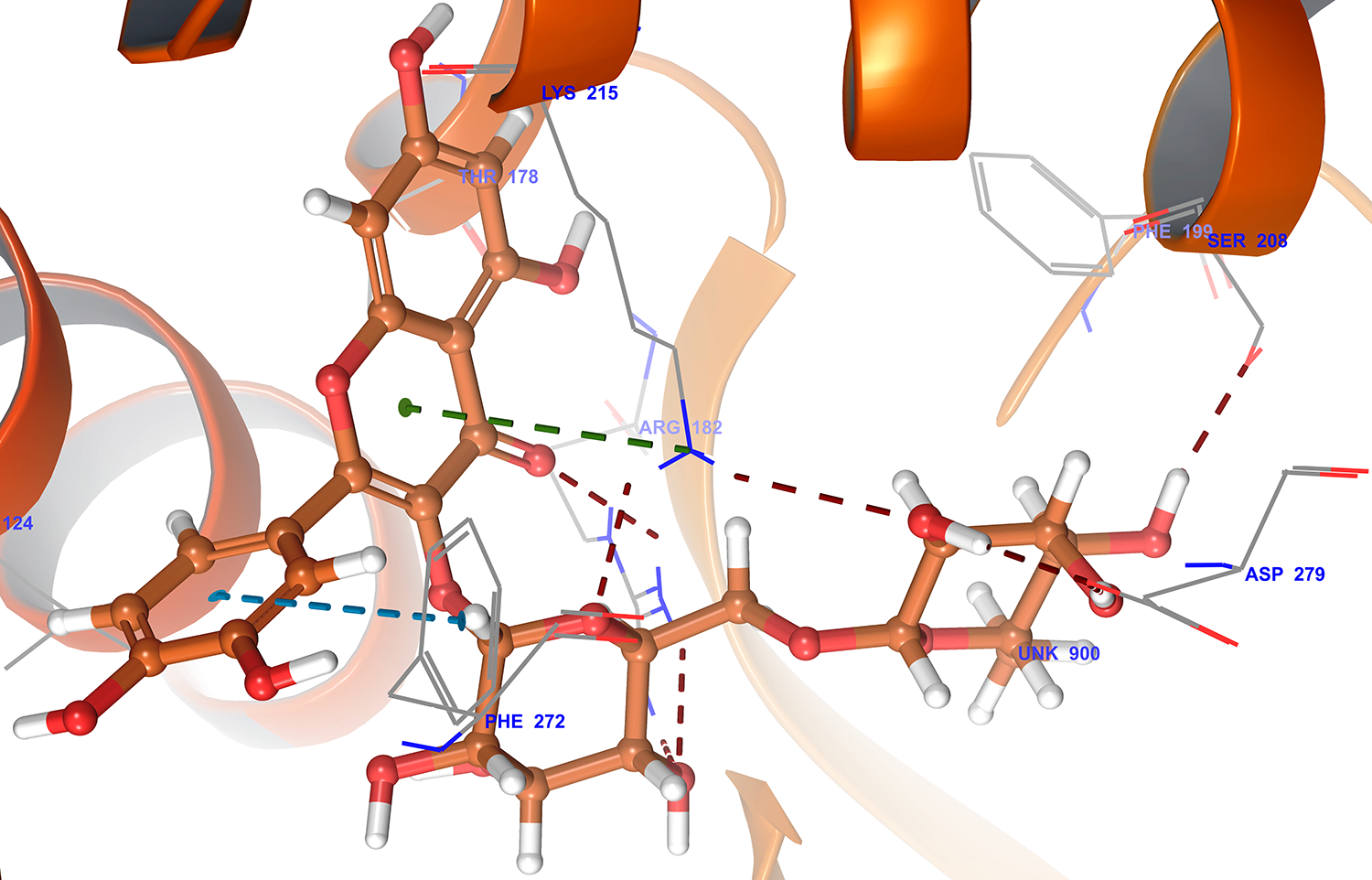
Molecular docking against ACE2
The results of the docking studies revealed that the compounds rutin, verbascoside, and hesperidin bound tightly at the active site of ACE2. The compound rutin (docking score: -11.5 kcal/mol) well occupied the receptor cavity through hydrogen bonding withAla348, Asp350, Phe390, Ser44, Ser47Arg393, Asn394 and verbascoside (docking score: -10.2 kcal/mol) showed hydrogen bonding interaction withAla348,Asp350, Asp382, Phe390, Glu375, His37, Asn394, and Glu402,whereas hesperidin (docking score: -9.5 kcal/mol) showed hydro-gen bonding interaction withAla348, Asp350,Trp69, and Tyr385 (Table 1).
 Molecular docking against RdRp of SARS-Cov-2
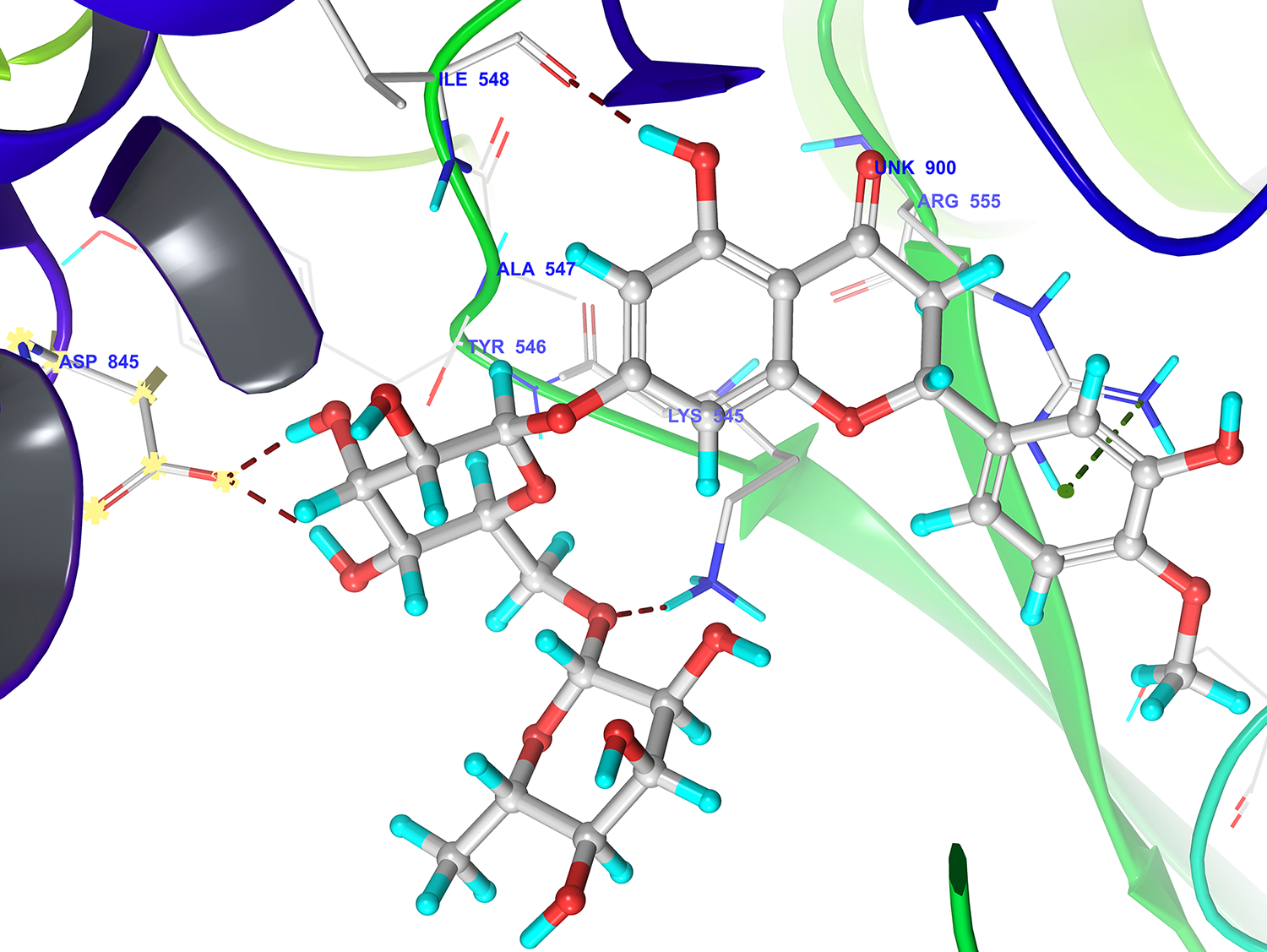
Molecular docking against RdRp of SARS-Cov-2
Phytoconstituents to have better docking at the RdRp than remdesivir (- 6.7 kcal/mol), For example, hesperidin (docking score -10.01) bound tightly in the active site of RdRp. Hesperidin well occupied in the receptor cavity and makes hydrogen bonding with Arg555, ILE548, Asp845, and Lys545. The phytoconstituents provide promising lead compounds for designing novel RdRp inhibitors.
Molecular docking against ORF8
Molecular docking studies were performed against ORF8 and results reveal that the compounds luteolin-O-glucoside and hesperidin bound tightly to an active site of ORF8. The compounds luteolin-O-glucoside and hesperidin interacts with Tyr42,Phe43, His58, Lys61, His87 and Asn97. These compounds showed docking scores of -13.06 and -14.45 kcal/mol, respectively, against ORF8 and provide insights into developing ORF8 inhibitors.
Docking studies revealed that phytoconstituents interact with ACE2. Although the functional implications of these phytoconstituents on ACE2 activity are yet to be confirmed, available evidence suggests that inhibition of ACE2 may be a pivotal step to inhibit SARS-CoV-2 virus infection and consequent inflammatory damage. Phytoconstituents may interact with catalytic and/or S-protein binding sites. Phytoconstituents also may have inhibitory actions on SARS-CoV-2 replication. Thus, phytoconstituents may have multiple sites of action, protecting and countering the inflammatory damage. Phytoconstituents may be useful both as prophylactic and treatment for COVID-19.
Conclusions
Currently, chemo informatics and molecular modelling method approaches are becoming vital components in the drug discovery due to reduction in cost and time. Computational methods signify a competent approach to efficiently alter large and diverse compound libraries to potential candidates for drug development. There are many compounds suggested by computational methods that could be evaluated quickly with in vitro techniques.
(The authors are from Delhi Pharmaceutical Sciences and Research University, New Delhi)